Metabarcoding
| Del av en serie om |
| DNA-streckkodning |
|---|

|
| By taxa |
| Övrig |
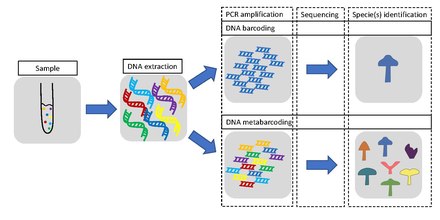
Metabarcoding är streckkodning av DNA / RNA (eller eDNA / eRNA ) på ett sätt som möjliggör samtidig identifiering av många taxa inom samma prov. Den största skillnaden mellan streckkodning och metabarcoding är att metabarcoding inte fokuserar på en specifik organism, utan syftar istället till att bestämma artsammansättningen inom ett prov.
En streckkod består av en kort variabel genregion (se till exempel olika markörer/streckkoder ) som är användbar för taxonomisk tilldelning flankerad av mycket konserverade genregioner som kan användas för primerdesign . Denna idé om allmän streckkodning har sitt ursprung 2003 från forskare vid University of Guelph .
Metabarcoding proceduren, liksom allmän streckkodning, fortsätter i ordning genom stadier av DNA-extraktion , PCR-amplifiering , sekvensering och dataanalys . Olika gener används beroende på om syftet är att streckkoda enstaka arter eller metabarcoding flera arter. I det senare fallet används en mer universell gen. Metabarcoding använder inte enstaka arter av DNA/RNA som utgångspunkt, utan DNA/RNA från flera olika organismer härrörande från ett miljö- eller bulkprov.
Miljö-DNA
Miljö-DNA eller eDNA beskriver det genetiska material som finns i miljöprover som sediment, vatten och luft, inklusive hela celler, extracellulärt DNA och potentiellt hela organismer. eDNA kan fångas från miljöprover och bevaras , extraheras , amplifieras , sekvenseras och kategoriseras baserat på dess sekvens. Från denna information är upptäckt och klassificering av arter möjlig. eDNA kan komma från hud, slemhinnor, saliv, spermier, sekret, ägg, avföring, urin, blod, rötter, löv, frukt, pollen och ruttnande kroppar från större organismer, medan mikroorganismer kan erhållas i sin helhet . eDNA-produktion är beroende av biomassa , ålder och matningsaktivitet samt fysiologi, livshistoria och rymdanvändning.
Till 2019 hade metoder inom eDNA-forskning utökats för att kunna bedöma hela samhällen från ett enda urval. Denna process involverar metabarcoding, som exakt kan definieras som användningen av allmänna eller universella polymeraskedjereaktions (PCR) primers på blandade DNA-prover från vilket ursprung som helst följt av nästa generations sekvensering med hög genomströmning (NGS) för att bestämma artsammansättningen av prov. Denna metod har varit vanlig inom mikrobiologi i åratal, men från och med 2020 har den bara hittat sin fot i bedömningen av makroorganismer. Ekosystemomfattande tillämpningar av eDNA-metabarcoding har potential att inte bara beskriva samhällen och biologisk mångfald, utan också att upptäcka interaktioner och funktionell ekologi över stora rumsliga skalor, även om det kan begränsas av falska avläsningar på grund av kontaminering eller andra fel. Sammantaget ökar eDNA-metabarcoding hastighet, noggrannhet och identifiering jämfört med traditionell streckkodning och minskar kostnaderna, men måste standardiseras och förenas, integrera taxonomi och molekylära metoder för fullständiga ekologiska studier.


eDNA-metabarcoding har tillämpningar för mångfaldsövervakning över alla livsmiljöer och taxonomiska grupper, forntida ekosystemrekonstruktion, växt-pollinator-interaktioner, kostanalys, upptäckt av invasiva arter, föroreningssvar och övervakning av luftkvalitet. eDNA-metabarcoding är en unik metod som fortfarande är under utveckling och kommer sannolikt att förbli i förändring under en tid när tekniken går framåt och procedurerna blir standardiserade. Men eftersom metabarcoding optimeras och dess användning blir mer utbredd, kommer det sannolikt att bli ett viktigt verktyg för ekologisk övervakning och globala bevarandestudier.
Gemenskapens DNA
Sedan starten av HTS (High-Throughput Sequencing ) har användningen av metabarcoding som ett verktyg för upptäckt av biologisk mångfald rönt ett enormt intresse. Det har dock ännu inte funnits klarhet angående vilket källmaterial som används för att utföra metabarcoding-analyser (t.ex. miljö-DNA kontra gemenskaps-DNA ). Utan klarhet mellan dessa två källmaterial kan skillnader i provtagning, såväl som skillnader i laboratorieförfaranden, påverka efterföljande bioinformatikpipelines som används för databehandling och komplicera tolkningen av rumsliga och tidsmässiga biologiska mångfaldsmönster. Här försöker vi tydligt skilja mellan de rådande källmaterialen som används och deras effekt på nedströmsanalys och tolkning för miljö-DNA-metabarcoding av djur och växter jämfört med gemenskaps-DNA-metabarcoding.
Med gemenskaps-DNA-metabarcoding av djur och växter, samlas målgrupperna oftast in i bulk (t.ex. jord, sjukdomsfälla eller nät), och individer avlägsnas från andra provskräp och slås ihop innan bulk-DNA-extraktion. Däremot isoleras makroorganism eDNA direkt från ett miljömaterial (t.ex. jord eller vatten) utan föregående segregering av enskilda organismer eller växtmaterial från provet och antar implicit att hela organismen inte finns i provet. Naturligtvis kan gemenskaps-DNA-prover innehålla DNA från delar av vävnader, celler och organeller från andra organismer (t.ex. tarminnehåll, kutant intracellulärt eller extracellulärt DNA). Likaså kan eDNA-prover från makroorganismer oavsiktligt fånga hela mikroskopiska icke-målorganismer (t.ex. protister, bakterier). Därmed kan distinktionen åtminstone delvis bryta ner i praktiken.
En annan viktig skillnad mellan gemenskaps-DNA och makroorganism-eDNA är att sekvenser som genereras från gemenskaps-DNA-metabarcoding kan verifieras taxonomiskt när proverna inte förstörs i extraktionsprocessen. Här kan sekvenser sedan genereras från voucherprover med hjälp av Sanger-sekvensering. Eftersom proverna för eDNA-metabarcoding saknar hela organismer kan inga sådana in situ-jämförelser göras. Taxonomiska affiniteter kan därför endast fastställas genom att direkt jämföra erhållna sekvenser (eller genom bioinformatiskt genererade operationella taxonomiska enheter (MOTU)), med sekvenser som är taxonomiskt annoterade såsom NCBI:s GenBank nukleotiddatabas, BOLD , eller med självgenererade referensdatabaser från Sanger‐ sekvenserat DNA. (Den molekylära operationella taxonomiska enheten (MOTU) är en grupp som identifieras genom användning av klusteralgoritmer och en fördefinierad procentuell sekvenslikhet, till exempel 97%)). Sedan, för att åtminstone delvis bekräfta den resulterande listan över taxa, görs jämförelser med konventionella fysiska, akustiska eller visuellt baserade undersökningsmetoder som utförs samtidigt eller jämförs med historiska register från undersökningar för en plats (se tabell 1).
Skillnaden i källmaterial mellan gemenskaps-DNA och eDNA har därför distinkta förgreningar för att tolka skalan av slutsatser för tid och rum om den biologiska mångfalden som upptäckts. Från samhällets DNA är det tydligt att de enskilda arterna hittades i den tid och plats, men för eDNA kan organismen som producerade DNA:t vara uppströms från provtagningsplatsen, eller så kan DNA:t ha transporterats i avföringen från en mer mobila rovdjursarter (t.ex. fåglar som deponerar fisk eDNA, eller var tidigare närvarande, men inte längre aktiva i samhället och upptäckt är från DNA som fälldes år till årtionden tidigare. Det senare betyder att skalan av slutledning både i rum och tid måste övervägas noggrant när man sluter sig till förekomsten för arten i samhället baserat på eDNA.
Metabarcoding stadier

Det finns sex stadier eller steg i DNA-streckkodning och metabarcoding. DNA-streckkodningen för djur (och specifikt för fladdermöss ) används som exempel i diagrammet till höger och i diskussionen omedelbart nedan.
Först väljs lämpliga DNA-streckkodande regioner för att svara på någon specifik forskningsfråga. Den vanligaste DNA-streckkodsregionen för djur är ett segment som är cirka 600 baspar långt av den mitokondriella genen cytokromoxidas I ( CO1). Detta lokus ger stor sekvensvariation mellan arter men ändå relativt liten mängd variation inom arter. Andra vanliga streckkodsregioner som används för artidentifiering av djur är ribosomala DNA (rDNA) regioner såsom 16S , 18S och 12S och mitokondriella regioner såsom cytokrom B. Dessa markörer har fördelar och nackdelar och används för olika ändamål. Längre streckkodsregioner (minst 600 baspar långa) behövs ofta för noggrann artavgränsning, särskilt för att särskilja nära släktingar. Identifiering av producenten av organismlämningar såsom avföring, hårstrån och saliv kan användas som ett proxymått för att verifiera frånvaro/närvaro av en art i ett ekosystem. DNA i dessa lämningar är vanligtvis av låg kvalitet och kvantitet, och därför används kortare streckkoder på cirka 100 baspar långa i dessa fall. På samma sätt bryts DNA-rester i dynga ofta också ned, så korta streckkoder behövs för att identifiera byten som konsumerats.
För det andra måste en referensdatabas byggas upp av alla DNA-streckkoder som kan förekomma i en studie. Helst behöver dessa streckkoder genereras från kupongexemplar som deponerats på en allmänt tillgänglig plats, som till exempel ett naturhistoriskt museum eller annat forskningsinstitut. Att bygga upp sådana referensdatabaser pågår för närvarande över hela världen. Partnerorganisationer samarbetar i internationella projekt som International Barcode of Life Project (iBOL) och Consortium for the Barcode of Life (CBOL), i syfte att konstruera en DNA-streckkodsreferens som kommer att ligga till grund för DNA-baserad identifiering av världens biom. Välkända streckkodsförråd är NCBI GenBank och Barcode of Life Data System (BOLD).
För det tredje måste cellerna som innehåller DNA av intresse brytas upp för att exponera dess DNA. Detta steg, DNA-extraktioner och reningar , bör utföras från substratet som undersöks. Det finns flera förfaranden tillgängliga för detta. Specifika tekniker måste väljas för att isolera DNA från substrat med delvis nedbrutet DNA, till exempel fossila prover, och prover som innehåller inhibitorer, såsom blod, avföring och jord. Extraktioner där DNA-utbytet eller kvaliteten förväntas vara låg bör utföras i en gammal DNA- anläggning, tillsammans med etablerade protokoll för att undvika kontaminering med modernt DNA. Experiment bör alltid utföras i duplikat och med positiva kontroller inkluderade.
För det fjärde måste amplikoner genereras från DNA som extraherats, antingen från ett enstaka prov eller från komplexa blandningar med primrar baserade på DNA-streckkoder valda under steg 1. För att hålla reda på deras ursprung måste märkta nukleotider (molekylära ID eller MID-märkningar) vara läggs till vid metabarcoding. Dessa etiketter behövs senare i analyserna för att spåra avläsningar från en bulkdatauppsättning tillbaka till deras ursprung.

För det femte bör lämpliga tekniker väljas för DNA-sekvensering . Den klassiska Sanger-kedjetermineringsmetoden bygger på selektiv inkorporering av kedjeförlängande hämmare av DNA-polymeras under DNA-replikation . Dessa fyra baser separeras efter storlek med hjälp av elektrofores och identifieras senare genom laserdetektering. Sangermetoden är begränsad och kan producera en enda läsning samtidigt och är därför lämplig för att generera DNA-streckkoder från substrat som bara innehåller en enda art. Nya teknologier som nanopore-sekvensering har resulterat i att kostnaden för DNA-sekvensering har minskat från cirka 30 000 USD per megabyte 2002 till cirka 0,60 USD 2016. Moderna nästa generations sekvenseringsteknologier (NGS) kan hantera tusentals till miljoner läsningar parallellt och är därför lämplig för massidentifiering av en blandning av olika arter som finns i ett substrat, sammanfattat som metabarcoding.
Slutligen måste bioinformatiska analyser utföras för att matcha DNA-streckkoder som erhållits med streckkodsindexnummer (BIN) i referensbibliotek. Varje BIN, eller BIN-kluster, kan identifieras till artnivå när den visar hög (>97 %) överensstämmelse med DNA-streckkoder kopplade till en art som finns i ett referensbibliotek, eller när taxonomisk identifiering till artnivån fortfarande saknas, en operationell taxonomic unit (OTU), som hänvisar till en grupp arter (dvs. släkte, familj eller högre taxonomisk rangordning). (Se binning (metagenomics) ). Resultaten av bioinformatikpipelinen måste beskäras, till exempel genom att filtrera bort opålitliga singletons, överflödiga dubbletter, lågkvalitetsläsningar och/eller chimära läsningar . Detta görs vanligtvis genom att utföra seriella BLAST-sökningar i kombination med automatisk filtrering och trimning av skript. Standardiserade trösklar behövs för att skilja mellan olika arter eller en korrekt och en felaktig identifiering.
Metabarcoding arbetsflöde
Trots den uppenbara kraften i tillvägagångssättet påverkas eDNA-metabarcoding av precisions- och noggrannhetsutmaningar fördelade över hela arbetsflödet på fältet, i laboratoriet och på tangentbordet. Som framgår av diagrammet till höger, efter den inledande studiedesignen (hypotes/fråga, målgrupp, etc.) består det nuvarande eDNA-arbetsflödet av tre komponenter: fält, laboratorium och bioinformatik. Fältkomponenten består av provtagning (t.ex. vatten, sediment, luft) som konserveras eller fryses innan DNA-extraktion. Laboratoriekomponenten har fyra grundläggande steg: (i) DNA koncentreras (om det inte utförs i fält) och renas, (ii) PCR används för att amplifiera en målgen eller målregion, (iii) unika nukleotidsekvenser som kallas "index" ( även kallade "streckkoder") inkorporeras med PCR eller ligeras ( bundna) till olika PCR-produkter, vilket skapar ett "bibliotek" där flera prover kan slås samman, och (iv) poolade bibliotek sekvenseras sedan med hög genomströmning maskin . Det sista steget efter laboratoriebehandling av prover är att beräkningsmässigt bearbeta utdatafilerna från sekvenseraren med hjälp av en robust bioinformatikpipeline.


OTU:er och artbegreppet
Metod och visualisering

a) Alfadiversitet visas som taxonomistapeldiagram, som visar relativ mängd taxa över prover med hjälp av Phinch-datavisualiseringsramverket (Bik & Pitch Interactive 2014). b) Beta-diversitetsmönster illustrerade via Principal Coordinate Analyser utförda i QIIME , där varje punkt representerar ett prov och färger särskiljer olika klasser av prov. Ju närmare två provpunkter i 3D-rymden, desto mer lika deras gemenskapssammansättningar. c) GraPhalAn fylogenetisk visualisering av miljödata, med cirkulära värmekartor och överflödsstaplar som används för att förmedla kvantitativa taxonegenskaper. d) Edge PCA, ett trädbaserat mångfaldsmått som identifierar specifika linjer (gröna/orange grenar) som bidrar mest till samhällsförändringar som observerats i prover fördelade över olika PCA-axlar.
Metoden kräver att varje insamlat DNA arkiveras med sitt motsvarande "typprov" (ett för varje taxon), utöver de vanliga insamlingsdata. Dessa typer lagras i specifika institutioner (museer, molekylära laboratorier, universitet, zoologiska trädgårdar, botaniska trädgårdar, herbarier, etc.) en för varje land, och i vissa fall är samma institution tilldelad att innehålla typer av mer än ett land , i fall där vissa nationer inte har teknologin eller ekonomiska resurser för att göra det.
På detta sätt representerar skapandet av typexemplar av genetiska koder en metodik parallell med den som utförs av traditionell taxonomi.
I ett första steg definierades den region av DNA som skulle användas för att göra streckkoden. Den måste vara kort och uppnå en hög andel unika sekvenser. För djur, alger och svampar har en del av en mitokondriell gen som kodar för subenhet 1 av cytokromoxidasenzymet CO1 gett höga procentandelar (95 %), en region runt 648 baspar.
När det gäller växter har användningen av CO1 inte varit effektiv eftersom de har låga nivåer av variation i den regionen, förutom de svårigheter som orsakas av de frekventa effekterna av polyploidi, introgression och hybridisering , så kloroplastgenomet verkar mer passande .
Ansökningar
Pollinatornätverk

↑ metabarcoding ↑ besöksundersökningar (a,b) växtpollinatorgrupper (c,d) växtpollinatorarter (e,f) individuella pollinator-växtarter ( Empis leptempis pandellei )
Apis: Apis mellifera ; Bomb.: Bombus sp.; W.bee: vilda bin; O.Hym.: andra Hymenoptera ; O.Dipt.: Andra Diptera ; Emp.: Empididae ; Syrph.: Syrphidae ; Överste: Coleoptera ; Lep.: Lepidoptera ; Musk.: Muscidae . Linjetjockleken framhäver andelen interaktioner
Diagrammet till höger visar en jämförelse av pollineringsnätverk baserat på DNA-metabarcoding med mer traditionella nätverk baserat på direkta observationer av insektsbesök på växter. Genom att upptäcka många ytterligare dolda interaktioner förändrar metabarcoding data till stor del egenskaperna hos pollineringsnätverken jämfört med besöksundersökningar. Molekylära data visar att pollinatörer är mycket mer generalister än förväntat från besöksundersökningar. Emellertid bestod pollinatörarter av relativt specialiserade individer och bildade funktionella grupper mycket specialiserade på blommorfer .
Som en konsekvens av de pågående globala förändringarna har en dramatisk och parallell världsomspännande minskning av pollinatörer och djurpollinerade växtarter observerats. Att förstå pollineringsnätverkens svar på dessa nedgångar är brådskande för att diagnostisera de risker som ekosystemen kan medföra samt för att utforma och utvärdera effektiviteten av bevarandeåtgärder. Tidiga studier av djurpollinering handlade om förenklade system, dvs specifika parvisa interaktioner eller involverade små undergrupper av växt-djursamhällen. Emellertid sker effekterna av störningar genom mycket komplexa interaktionsnätverk och numera är dessa komplexa system för närvarande ett stort forskningsfokus. Att bedöma de verkliga nätverken (bestämda av ekologisk process) från fältundersökningar som är föremål för provtagningseffekter ger fortfarande utmaningar.
Nyare forskningsstudier har tydligt dragit nytta av nätverkskoncept och verktyg för att studera interaktionsmönstren i stora artsammansättningar. De visade att växtpollinatornätverk var mycket strukturerade och avviker avsevärt från slumpmässiga associationer. Vanligtvis har nätverk (1) en låg anslutning (den realiserade andelen av alla potentiella länkar i samhället) vilket tyder på en låg grad av generalisering; (2) en hög kapsling (ju mer specialiserade organismer är mer benägna att interagera med undergrupper av arterna som mer generalistiska organismer interagerar med) desto mer specialiserade arter interagerar endast med korrekta undergrupper av dessa arter som interagerar med de mer generalistiska; (3) en kumulativ fördelning av anslutningsmöjligheter (antal länkar per art, s) som följer en makt eller en trunkerad maktlagsfunktion som kännetecknas av få supergeneralister med fler länkar än förväntat av en slump och många specialister; (4) en modulär organisation. En modul är en grupp av växt- och pollinatörarter som uppvisar höga nivåer av anslutning inom modulen och som är dåligt kopplade till arter av andra grupper.
Den låga anslutningsgraden och den höga andelen specialister inom pollineringsnätverk står i kontrast till uppfattningen att generalisering snarare än specialisering är normen i nätverk. De flesta växtarter besöks faktiskt av en mångfald av pollinatörer som utnyttjar blomresurser från ett brett spektrum av växtarter. En huvudorsak som framkallats för att förklara denna uppenbara motsägelse är det ofullständiga urvalet av interaktioner. De flesta nätverksegenskaper är faktiskt mycket känsliga för samplingsintensitet och nätverksstorlek. Nätverksstudier är i grunden fytocentriska, dvs baserade på observationer av pollinatörsbesök på blommor. Detta växtcentrerade tillvägagångssätt lider ändå av inneboende begränsningar som kan hämma förståelsen av mekanismer som bidrar till samhällsförsamling och biologiska mångfaldsmönster. För det första är direkta observationer av pollinatörsbesök hos vissa taxa, såsom orkidéer, ofta sällsynta och sällsynta interaktioner är mycket svåra att upptäcka i fält i allmänhet. Pollinator- och växtsamhällen är vanligtvis sammansatta av ett fåtal arter och många sällsynta arter som är dåligt registrerade i besöksundersökningar. Dessa sällsynta arter framstår som specialister, medan de i själva verket kan vara typiska generalister. På grund av det positiva sambandet mellan interaktionsfrekvens (f) och anslutning (s), kan undersamplade interaktioner leda till att graden av specialisering i nätverk överskattas. För det andra har nätverksanalyser mestadels verkat på artnivåer. Nätverk har mycket sällan skalats upp till de funktionella grupperna eller nedskalats till de individbaserade nätverken, och de flesta av dem har fokuserats på endast en eller två arter. Beteendet hos antingen individer eller kolonier ignoreras vanligtvis, även om det kan påverka strukturen i artnätverken. Arter som redovisas som generalister i artnätverk kan därför innebära kryptiska specialiserade individer eller kolonier. För det tredje är blombesökare inte alltid effektiva pollinatörer eftersom de kanske inte deponerar något konspecifikt pollen och/eller mycket heterospecifikt pollen. Djurcentrerade tillvägagångssätt baserade på undersökningar av pollenbelastningar på besökare och växtstämpel kan vara mer effektiva när det gäller att avslöja interaktioner mellan växt och pollinatör.
Att lösa upp näringsnät

Metabarcoding erbjuder nya möjligheter för att dechiffrera trofiska kopplingar mellan rovdjur och deras byten i näringsnät. Jämfört med traditionella, tidskrävande metoder, såsom mikroskopiska eller serologiska analyser , möjliggör utvecklingen av DNA-metabarcoding identifiering av bytesarter utan förkunskaper om rovdjurens bytesområden. Dessutom kan metabarcoding också användas för att karakterisera ett stort antal arter i en enda PCR- reaktion, och för att analysera flera hundra prover samtidigt. Ett sådant tillvägagångssätt används i allt större utsträckning för att utforska den funktionella mångfalden och strukturen hos näringsnät i agroekosystem. Liksom andra molekylärbaserade tillvägagångssätt ger metabarcoding endast kvalitativa resultat på närvaron/frånvaron av bytesarter i tarmen eller fekala prover. Men denna kunskap om identiteten hos bytesdjur som konsumeras av rovdjur av samma art i en given miljö möjliggör ett "pragmatiskt och användbart surrogat för verkligt kvantitativ information.
Inom matnätsekologin är "vem som äter vem" en grundläggande fråga för att få en bättre förståelse av de komplexa trofiska interaktioner som finns mellan skadedjur och deras naturliga fiender inom ett givet ekosystem. Dietanalys av leddjur och ryggradsdjurs rovdjur gör det möjligt att identifiera nyckelrovdjur som är involverade i den naturliga kontrollen av leddjursskadegörare och ger insikter om bredden av deras diet ( generalist kontra specialist) och intraguild predation .
Diagrammet till höger sammanfattar resultat från en studie från 2020 som använde metabarcoding för att reda ut den funktionella mångfalden och strukturen hos näringsväven som är förknippad med ett par hirsfält i Senegal. Efter att ha tilldelats de identifierade OTU:erna som art identifierades 27 artropodbytesdjur från nio leddjursrovdjur. Det genomsnittliga antalet bytesdjur som detekterades per prov var det högsta hos karabidbaggar , myror och spindlar, och det lägsta bland de återstående rovdjuren, inklusive antokoride insekter, pentatomide insekter och örontvistar. Bland rovdjursleddjur observerades en stor mångfald av leddjursbyten hos spindlar, karabidbaggar, myror och antokoride insekter. Däremot var mångfalden av bytesarter som identifierats i örontvistar och pentatomida insekter relativt låg. Lepidoptera , Hemiptera , Diptera och Coleoptera var de vanligaste insekternas bytesdjur som upptäcktes från rovleddjur.
Att bevara funktionell biologisk mångfald och relaterade ekosystemtjänster , särskilt genom att bekämpa skadedjur med hjälp av sina naturliga fiender, erbjuder nya vägar för att tackla utmaningar för en hållbar intensifiering av livsmedelsproduktionssystem. Predation av skadedjur av grödor av generalistiska rovdjur, inklusive leddjur och ryggradsdjur, är en viktig del av naturlig skadedjursbekämpning . En särskilt viktig egenskap hos de flesta generalistiska rovdjur är att de kan kolonisera grödor tidigt på säsongen genom att först livnära sig på alternativa byten. Bredden av den "generalistiska" dieten medför dock vissa nackdelar för skadedjursbekämpning, såsom predation inom skrået. En avstämd diagnos av dietbredden hos generalistiska rovdjur, inklusive predation av bytesdjur som inte är skadegörare, behövs därför för att bättre reda ut näringsnäten (t.ex. exploateringskonkurrens och uppenbar konkurrens) och i slutändan för att identifiera nyckelfaktorer för naturlig skadedjursbekämpning i agroekosystem. Men betydelsen av generalistiska rovdjur i näringsväven är generellt sett svår att bedöma, på grund av den tillfälliga karaktären hos individuella interaktioner mellan rovdjur och bytesdjur. Det enda avgörande beviset på predation är resultatet av direkt observation av byteskonsumtion, identifiering av bytesrester i rovdjurens tarmar och analyser av uppstötningar eller avföring.
Marin biosäkerhet

.jpeg)


Spridningen av främmande arter (NIS) representerar betydande och ökande risker för ekosystemen. I marina system kan NIS som överlever transporten och anpassar sig till nya platser ha betydande negativa effekter på den lokala biologiska mångfalden, inklusive förskjutning av inhemska arter och förändringar i biologiska samhällen och tillhörande näringsnät. När NIS väl har etablerats är de extremt svåra och kostsamma att utrota, och ytterligare regional spridning kan ske genom naturlig spridning eller via antropogena transportvägar. Även om fartygsskrov och fartygs barlastvatten är välkända som viktiga antropogena vägar för den internationella spridningen av NIS, är jämförelsevis lite känt om potentialen hos regionalt transiterande fartyg att bidra till sekundär spridning av marina skadedjur genom länsvattentranslokation.
Nyligen genomförda studier har avslöjat att vattnet och tillhörande skräp som finns i länsutrymmen på små fartyg (<20 m) kan fungera som en vektor för spridning av NIS på regional skala. Länsvatten definieras som allt vatten som hålls kvar på ett fartyg (förutom ballast) och som inte avsiktligt pumpas ombord. Det kan samlas på eller under fartygets däck (t.ex. under golvpaneler) genom en mängd olika mekanismer, inklusive vågverkan, läckor, via propellerns akterkörtlar och genom lastning av föremål som dykning, fiske, vattenbruk eller vetenskaplig utrustning . Länsvatten kan därför innehålla såväl havsvatten som levande organismer i olika livsstadier, cellskräp och föroreningar (t.ex. olja, smuts, rengöringsmedel etc.), som alla vanligtvis töms ut med hjälp av automatiska länspumpar eller är självdränerande använda anknäbbsventiler. Länsvatten som pumpas från små fartyg (manuellt eller automatiskt) behandlas vanligtvis inte före utsläpp till havet, i motsats till större fartyg som krävs för att separera olja och vatten med hjälp av filtreringssystem, centrifugering eller kolabsorption. Om propagula är livskraftiga genom denna process, kan utsläpp av länsvatten resultera i spridning av NIS.
2017, Fletcher et al. använde en kombination av laboratorie- och fältexperiment för att undersöka mångfalden, förekomsten och överlevnaden av biologiskt material som finns i länsvattenprover tagna från små kustfartyg. Deras laboratorieexperiment visade att ascidiankolonier eller -fragment, och bryozoanlarver , kan överleva passage genom ett ofiltrerat pumpsystem i stort sett oskadda. De genomförde också den första morfomolekylära bedömningen (med hjälp av eDNA-metabarcoding) av biosäkerhetsrisken som utsläpps av länsvatten från 30 små fartyg (segelbåtar och motorbåtar) av olika ursprung och seglingstid. Med hjälp av eDNA-metabarcoding karakteriserade de ungefär tre gånger fler taxa än via traditionella mikroskopiska metoder, inklusive detektering av fem arter som erkänts som icke-inhemska i studieregionen.
För att hjälpa till att förstå riskerna förknippade med olika NIS-introduktionsvektorer, kompletteras traditionella biodiversitetsbedömningar av mikroskop i allt högre grad med eDNA-metabarcoding. Detta gör att ett brett spektrum av olika taxonomiska sammansättningar, i många livsstadier, kan identifieras. Det kan också möjliggöra upptäckt av NIS som kan ha förbisetts med traditionella metoder. Trots den stora potentialen hos eDNA-metabarcoding-verktyg för storskalig taxonomisk screening, är en nyckelutmaning för eDNA i samband med miljöövervakning av marina skadedjur, och särskilt när man övervakar slutna miljöer som vissa länsutrymmen eller ballasttankar, att skilja döda och livskraftiga organismer. Extracellulärt DNA kan finnas kvar i mörka/kalla miljöer under långa tidsperioder (månader till år, så många av de organismer som detekteras med eDNA-metabarcoding kanske inte har varit livskraftiga på platsen för provtagning på dagar eller veckor. Däremot kan ribonukleinsyra ( RNA) försämras snabbt efter celldöd, vilket sannolikt ger en mer exakt representation av livskraftiga samhällen. Nyligen genomförda metabarcoding-studier har utforskat användningen av samextraherade eDNA- och eRNA-molekyler för att övervaka bentiska sedimentprover runt marina fiskodlingar och oljeborrplatser, och har kollektivt hittade något starkare samband mellan biologiska och fysikalisk-kemiska variabler längs inverkansgradienter vid användning av eRNA. Ur en marin biosäkerhetsprospektiv kan upptäckten av levande NIS representera ett allvarligare och omedelbart hot än upptäckten av NIS enbart baserat på en DNA-signal. Miljö RNA kan därför erbjuda en användbar metod för att identifiera levande organismer i prover.
Diverse
Konstruktionen av det genetiska streckkodsbiblioteket var från början fokuserat på fiskar och fåglarna, som följdes av fjärilar och andra ryggradslösa djur. När det gäller fåglar tas DNA-provet vanligtvis från bröstet.
Forskare har redan utvecklat specifika kataloger för stora djurgrupper, som bin, fåglar, däggdjur eller fiskar. En annan användning är att analysera den fullständiga zoocenosen i ett givet geografiskt område, såsom projektet "Polar Life Bar Code" som syftar till att samla in de genetiska egenskaperna hos alla organismer som lever i polära områden; jordens båda poler. Relaterat till denna form är kodningen av all ichthyofauna i en hydrografisk bassäng, till exempel den som började utvecklas i Rio São Francisco, i nordöstra Brasilien .
Potentialen för användningen av streckkoder är mycket bred, eftersom upptäckten av många kryptiska arter (det har redan gett många positiva resultat), användningen för att identifiera arter i alla skeden av deras liv, den säkra identifieringen i fall av skyddade arter som är olagligt människohandel osv.
Potentialer och brister

Potentialer
DNA-streckkodning har föreslagits som ett sätt att särskilja arter som är lämpliga även för icke-specialister att använda.
Brister
I allmänhet gäller bristerna för DNA-streckkodning även för metabarcoding. En särskild nackdel för metabarcoding-studier är att det ännu inte finns någon konsensus om den optimala experimentella designen och bioinformatikkriterierna som ska tillämpas i eDNA-metabarcoding. Det finns dock för närvarande gemensamma försök, såsom COST-nätverket DNAqua-Net från European Cooperation in Science and Technology , för att gå framåt genom att utbyta erfarenheter och kunskaper för att etablera bästa praxisstandarder för bioövervakning.
Den så kallade streckkoden är en region av mitokondrie-DNA inom genen för cytokrom c-oxidas . En databas, Barcode of Life Data Systems (BOLD), innehåller DNA-streckkodssekvenser från över 190 000 arter. Men forskare som Rob DeSalle har uttryckt oro över att klassisk taxonomi och DNA-streckkodning, som de anser vara en felaktig benämning, måste förenas, eftersom de avgränsar arter på olika sätt. Genetisk introgression medierad av endosymbionter och andra vektorer kan ytterligare göra streckkoder ineffektiva vid identifiering av arter.
Status för streckkodsarter
Inom mikrobiologi kan gener röra sig fritt även mellan avlägset besläktade bakterier, eventuellt sträcka sig till hela bakteriedomänen. Som en tumregel har mikrobiologer antagit att typer av bakterier eller Archaea med 16S ribosomala RNA- gensekvenser som är mer lika varandra än 97 % behöver kontrolleras med DNA-DNA-hybridisering för att avgöra om de tillhör samma art eller inte. Detta koncept minskades 2006 till en likhet på 98,7 %.
DNA-DNA-hybridisering är föråldrad, och resultaten har ibland lett till missvisande slutsatser om arter, som med pomarin och stor skua . Moderna metoder jämför sekvenslikhet med beräkningsmetoder.
Se även
- Barcode of Life Data System (FET)
- Consortium for the Barcode of Life (CBOL)
- International Nucleotide Sequence Database Collaboration (INSDC)
- Molekylär markör
- Taxonomiska hinder
Ytterligare referenser
- ^ Santoferrara, Luciana; Burki, Fabien; Filker, Sabine; Logares, Ramiro; Dunthorn, Micah; McManus, George B. (2020). "Perspektiv från tio år av protistiska studier genom High-Throughput Metabarcoding". Journal of Eukaryotic Microbiology . 67 (5): 612–622. doi : 10.1111/jeu.12813 . hdl : 10261/223228 . PMID 32498124 . S2CID 219331807 .