Borylering
Metallkatalyserade C-H-boryleringsreaktioner är övergångsmetallkatalyserade organiska reaktioner som producerar en organoboronförening genom funktionalisering av alifatiska och aromatiska C-H-bindningar och är därför användbara reaktioner för aktivering av kol-vätebindningar . Metallkatalyserade C–H-boryleringsreaktioner använder övergångsmetaller för att direkt omvandla en C–H-bindning till en C–B-bindning. Denna väg kan vara fördelaktig jämfört med traditionella boryleringsreaktioner genom att använda billiga och rikliga kolväteutgångsmaterial, begränsa prefunktionaliserade organiska föreningar, minska giftiga biprodukter och effektivisera syntesen av biologiskt viktiga molekyler. Boronsyror och borsyraestrar är vanliga borylgrupper som införlivas i organiska molekyler genom boryleringsreaktioner. Boronsyror är trevärda borinnehållande organiska föreningar som har en alkylsubstituent och två hydroxylgrupper. På liknande sätt har borsyraestrar en alkylsubstituent och två estergrupper. Boronsyror och estrar klassificeras beroende på typen av kolgrupp (R) som är direkt bunden till bor, till exempel alkyl-, alkenyl-, alkynyl- och aryl-boronsyraestrar. Den vanligaste typen av utgångsmaterial som införlivar borsyraestrar i organiska föreningar för övergångsmetallkatalyserade boryleringsreaktioner har den allmänna formeln (RO) 2B -B(OR) 2 . Till exempel bis(pinacolato)dibor (B 2 Pin 2 ) och bis(catecholato) diboran (B 2 Cat 2 ) vanliga borkällor med denna allmänna formel.

Boratomen i en borsyraester eller borsyra är sp 2 hybridiserad med en ledig p-orbital, vilket gör att dessa grupper kan fungera som Lewis-syror . C–B-bindningen av borsyror och estrar är något längre än typiska C–C-enkelbindningar med ett intervall på 1,55-1,59 Å. Den förlängda C–B-bindningen i förhållande till C–C-bindningen resulterar i en bindningsenergi som också är något mindre än den för C–C-bindningar (323 kJ/mol för C–B mot 358 kJ/mol för C–C). Kol -vätebindningen har en bindningslängd på cirka 1,09 Å och en bindningsenergi på cirka 413 kJ/mol. C–B-bindningen är därför en användbar mellanprodukt som en bindning som ersätter en typiskt oreaktiv C–H-bindning.
Organoborföreningar är organiska föreningar som innehåller en kol-borbindning. Organoboronföreningar har breda tillämpningar för kemisk syntes eftersom C–B-bindningen lätt kan omvandlas till en C–X (X = Br, Cl), C–O, C–N eller C–C-bindning. På grund av C-B-bindningens mångsidighet har många processer utvecklats för att införliva dem i organiska föreningar. Organoboronföreningar syntetiseras traditionellt från Grignard-reagenser genom hydroborering eller diborationsreaktioner. Borylering ger ett alternativ.
Metallkatalyserade C–H-boryleringsreaktioner
Alifatisk CH-borylering
Som först beskrevs av Hartwig kan alkaner boryleras selektivt med hög selektivitet för den primära C–H-bindningen med användning av Cp*Rh(η 4 -C 6 Me 6 ) som katalysator. Noterbart är selektiviteten för den primära C-H-bindningen exklusiv även i närvaro av heteroatomer i kol-vätekedjan. Den rodiumkatalyserade boryleringen av metyl-CH-bindningar sker selektivt utan beroende av heteroatomens position. Borylering sker selektivt vid den minst steriskt hindrade och minst elektronrika primära C-H-bindningen i en rad acetaler , etrar , aminer och alkylfluorider. Dessutom har ingen reaktion visat sig ske i frånvaro av primära C–H-bindningar, till exempel när cyklohexan är substratet.

Selektiv funktionalisering av en primär alkanbindning beror på bildningen av ett kinetiskt och termodynamiskt gynnsamt primärt alkyl-metallkomplex över bildning av ett sekundärt alkyl-metallkomplex.

Den större stabiliteten hos primära kontra sekundära alkylkomplex kan tillskrivas flera faktorer. För det första gynnas det primära alkylkomplexet steriskt framför det sekundära alkylkomplexet. För det andra är partiella negativa laddningar ofta närvarande på a-kolet i ett metall-alkylkomplex och en primär alkylligand stödjer en partiell negativ laddning bättre än en sekundär alkylligand. Ursprunget till selektivitet för alifatisk C-H-borylering med rodiumkatalysatorer undersöktes med en typ av mekanistisk studie som kallas väte-deuteriumutbyte . H/D-utbyte visade att regioselektivitet för den övergripande processen som visas nedan är resultatet av selektiv klyvning av primära över sekundära C-H-bindningar och selektiv funktionalisering av den primära metall-alkyl-intermediären över den sekundära metall-alkyl-mellanprodukten.

Den syntetiska användbarheten av alifatisk C-H-borylering har tillämpats på modifiering av polymerer genom borylering följt av oxidation för att bilda hydroxylfunktionaliserade polymerer.

Aromatisk C–H borylering
Sterisk riktad CH-borylering av arener
Det första exemplet på en katalytisk C-H-borylering av ett oaktiverat kolväte (bensen) rapporterades av Smith och Iverson med användning av Ir(Cp*)(H)(Bpin) som katalysator. Verkningsgraden för detta system var dock låg och gav endast 3 omgångar efter 120 timmar vid 150 °C. Många efterföljande utvecklingar av Hartwig och medarbetare ledde till effektiva, praktiska förutsättningar för arenborylering. Aromatisk C-H-borylering utvecklades av John F. Hartwig och Ishiyama med användning av diborreagenset Bis(pinacolato)diboron katalyserad av 4,4'-di-tert-butylbipyridin (dtbpy) och [Ir(COD)(OMe)] 2 . Med detta katalysatorsystem sker boryleringen av aromatiska C-H-bindningar med regioselektivitet som styrs av steriska effekter av startarenen. Selektiviteten för funktionalisering av aromatiska C–H-bindningar styrs av den allmänna regeln att reaktionen inte sker orto till en substituent när en C–H-bindning som saknar en ortosubstituent är tillgänglig. När endast en funktionell grupp är närvarande sker borylering i meta- och parapositionen i statistiska förhållanden på 2:1 (meta:para). Orto - isomeren detekteras inte på grund av de steriska effekterna av substituenten.

Addition av Bpin sker endast i en position för symmetriskt substituerade 1,2- och 1,4-substituerade arener. Symmetriska eller osymmetriska 1,3-substituerade arener är också selektivt borylerade eftersom endast en C-H-bindning är steriskt tillgänglig.

Detta i motsats till elektrofil aromatisk substitution där regioselektivitet styrs av elektroniska effekter.

Den syntetiska betydelsen av aromatisk C-H-borylering visas nedan, där en 1,3-disubstituerad aromatisk förening direkt kan omvandlas till en 1,3,5-organoboranförening och därefter funktionaliseras.

Aromatisk CH-funktionalisering införlivades framgångsrikt i den totala syntesen av Complanadine A, en Lycopodium -alkaloid som förbättrar mRNA- uttryck för nervtillväxtfaktor (NGF) och produktionen av NGF i mänskliga gliaceller . Naturliga produkter som främjar tillväxten av nya neurala nätverk är av intresse vid behandling av sjukdomar som Alzheimers sjukdom . Complanadine A syntetiserades framgångsrikt med en kombination av direkt aromatisk C-H-borylering utvecklad av Hartwig och Ishyiama, följt av Suzuki-Miyaura-korskoppling , sedan klyvning av Boc-skyddsgruppen .

C–H-borylering av heteroarener
Heteroarener kan också genomgå borylering under iridiumkatalyserade förhållanden, men platsselektiviteten i detta fall styrs av elektroniska effekter , där furaner , pyrroler och tiofener genomgår reaktion vid CH-bindningen alfa till heteroatomen. I detta fall föreslås selektivitet ske genom C–H-bindningen alfa till heteroatomen eftersom det är den suraste C–H-bindningen och därför den mest reaktiva.

Riktad orto C–H borylering
Med användning av samma katalysatorsystem kan styrande grupper användas för att uppnå regioselektivitet utan substituenter som steriska mediatorer. Till exempel rapporterade Boebel och Hartwig en metod för att utföra orto -borylering där en dimetyl-hydrosilyl-riktande grupp på arenen genomgår iridiumkatalyserad borylering vid C-H-bindningen orto till den silanstyrande gruppen. Selektivitet för ortopositionen vid användning av hydrosilylriktande grupper har tillskrivits reversibel addition av Si-H-bindningen till metallcentrum, vilket leder till preferentiell klyvning av C-H-bindningen orto till hydrosilylsubstituenten. Flera andra strategier för att uppnå orto -borylering av arener har utvecklats med användning av olika styrgrupper.

Mekanistisk detalj för C–H borlyation av arenor
Ett trisboryliridiumkomplex har föreslagits för att underlätta mekanismen för var och en av dessa reaktioner som resulterar i C-H-borylering av arener och heteroarener. Kinetiska studier och isotopmärkningsstudier har visat att ett Ir(III) -triborylkomplex reagerar med arenet i den katalytiska processen. En version av den katalytiska cykeln visas nedan för ortoborylering av hydrosilanföreningar. Kinetiska data visar att ett observerat trisborylkomplex koordinerat till cyklookten snabbt och reversibelt dissocierar cyklookten för att bilda ett trisborylkomplex med 16 elektroner. I fallet med användning av bensyldimetylsilan som en styrgrupp föreslås det att bensyldimetylsilan reagerar med trisboryliridiumkatalysatorn genom reversibel addition av Si-H-bindningen till metallcentret, följt av selektiv orto-C–H-bindningsaktivering via oxidativ addition och reduktiv eliminering .

Meta-selektiv borylering : Meta-selektiv C-H borylering är en viktig syntetisk transformation, som upptäcktes 2002 av Smith III från Michigan State University, USA. Emellertid var denna metaborylering fullständigt steriskt riktad och begränsades till endast 1,3-disubstituerade bensener. Omkring 12 år senare upptäckte Dr. Chattopadhyay och hans team från Center of Biomedical Research, UP, Indien en elegant teknologi för den meta-selektiva aktiveringen och boryleringen av C–H-bindningar. Teamet hade visat att med samma substrat kan man byta den andra positionsselektiviteten genom att bara ändra liganden. Ursprunget för meta-selektiviteten definierades av de två parametrarna, såsom: 1) elektrostatisk interaktion, 2) en sekundär BN-interaktion.
Samtidigt rapporterade ett team från Japan, Dr. Kanai, ett fantastiskt koncept för den meta-selektiva boryleringen baserat på den sekundära interaktionen. Denna metod omfattar olika borylering av karbonylföreningar.
Reduktionsreaktioner med organoboronföreningar
Corey–Bakshi–Shibata-reduktion (CBS-reduktion)
1981 har Hirao och medarbetare funnit att asymmetrisk reduktion av prokirala aromatiska ketoner med kirala aminoalkoholer och boran gav motsvarande sekundära alkoholer med 60 % ee . De fick reda på att de kirala aminoalkoholerna skulle reagera med boran för att bilda aloxylamin-borankomplex. Komplexen föreslås innehålla ett relativt styvt system med fem medlemmar, vilket gör dem termiskt och hydrolytiskt stabila och lösliga i en mängd olika protiska och aprotiska lösningsmedel.
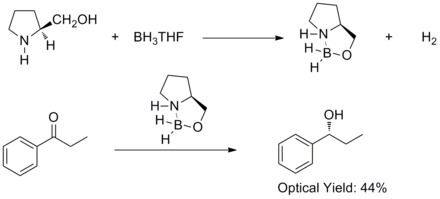
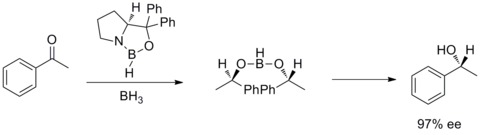
År 1987 upptäckte Elias James Corey och hans medarbetare att bildandet av oxazaborolidiner från boran och kirala aminoalkoholer. Och oxazaborolidinerna visade sig katalysera den snabba och mycket enantioselektiva reduktionen av prokirala ketoner i närvaro av BH3THF. Denna enantioselektiva reduktion av akirala ketoner med katalytisk oxazaborolidin kallas Corey-Bakshi-Shibata-reduktion eller CBS-reduktion.
Midland Alpine-boran-reduktion (Midland-reduktion)
1977 rapporterade MM Midland och medarbetare en överraskande observation att B-3-alfa-Pinanyl-9-borabicyklo[3,3,1]-nonan, lätt framställd genom hydroborering av (+)-alfa-pinen med 9- borobicyklo [3,3,1]nonan reducerar snabbt bensaldehyd-alfa-d till (S)-(+)-bensyl-alfa-d alkohol med en väsentligen kvantitativ asymmetrisk induktion.
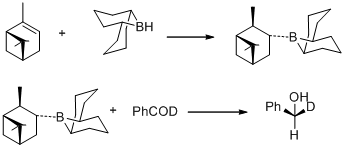
Samma år upptäckte MM Midland B-3-alfa-pinanyl-9-BBN som reduktionsmedel, vilket kunde vara lätt tillgängligt genom att reagera (+)-alfa-pinen med 9-BBN. Det nya reduktionsmedlet kommersialiserades senare av Aldrich Co. under namnet Alpine Borane och den asymmetriska reduktionen av karbonylgrupper med endera enantiomer av Alpine-Borane är känd som Midland Alpine-Borane-reduktion.
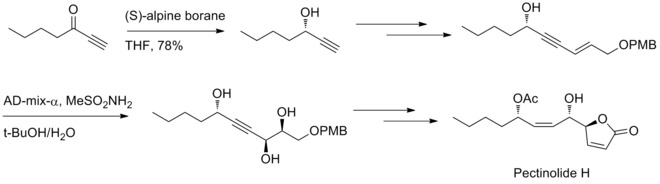
Under 2012 har URY Venkateswarlu och medarbetare rapporterat en stereoselektiv metod för att syntetisera pektinolid H. Midland-reduktion och Sharpless dihydroxyleringsreaktion är involverade i genereringen av de tre kirala centran vid C–4', C–5 och C–1'.
Kopplingsreaktioner med organoboronföreningar
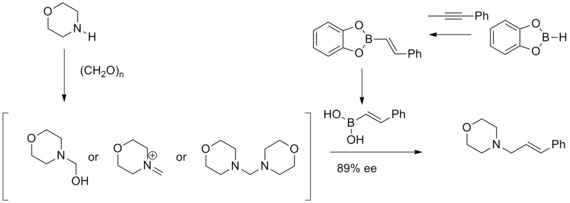
Petasis boronsyra-Mannich reaktion
1993 rapporterade NA Petasis och I. Akrltopoulou en effektiv syntes av allyliska aminer med en modifierad Mannich-reaktion . I denna modifierade Mannich-reaktion har de funnit att vinylboronsyror kan delta som nukleofiler för att ge geometriskt rena allylaminer. Denna modifierade Mannich-reaktion var känd som Petasis boronsyra-Mannich-reaktion.
Roush asymmetrisk allylering
1978 rapporterade RW Hoffmann och T. Herold om den enantioselektiva syntesen av sekundära homoallylalkoholer via kirala icke-racemiska allylboronsyraestrar . De homoallyliska alkoholerna bildades med utmärkt utbyte och måttlig enantioselektivitet.
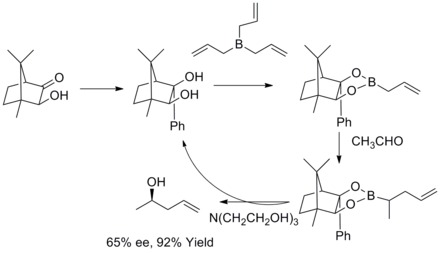
1985 upptäckte WR Roush och medarbetare att tartratmodifierade allylboronater erbjuder ett enkelt, mycket attraktivt tillvägagångssätt för kontroll av ansiktsselektivitet i reaktioner med kirala och akirala aldehyder. Under de följande åren utökade WR Roush och medarbetare denna strategi till syntesen av but-2-en-1,4-dioler och antidioler. Denna typ av reaktion är känd som Rouch asymmetrisk allylering.
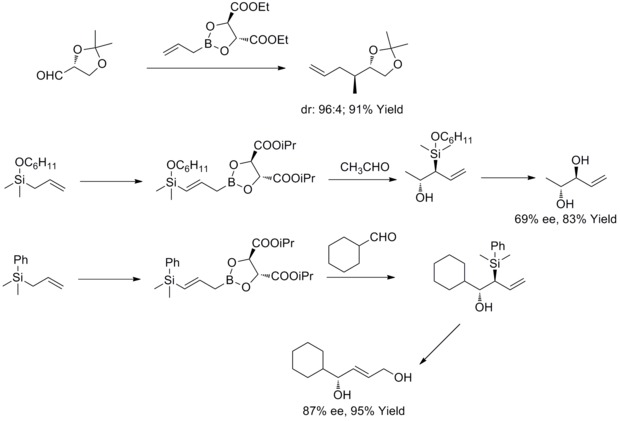
Under 2011 har RA Fernandes och P. Kattanguru slutfört en förbättrad total syntes av (8S, 11R, 12R)- och (8R, 11R, 12R)-topsentolid B2 diastereomerer i åtta steg. I uppsatsen användes diastereoselektiv Roush-allyleringsreaktion som en nyckelreaktion i den totala syntesen för att introducera två kirala mellanprodukter. Och sedan syntetiserade författarna de två diastereomererna genom dessa två kirala mellanprodukter.

Suzuki–Miyaura korskoppling
1979 rapporterade N. Miyaura och A. Suzuki syntesen av arylerade (E)-alkener i högt utbyte från arylhalider med alkyl-1-enylboraner och katalyserad av tetrakis( trifenylfosfin )palladium och baser. Sedan utökar A. Suzuki och medarbetare denna typ av reaktion till andra organoboronföreningar och andra alkenyl, aryl , alkylhalider och triflat . De palladiumkatalyserade organoborföreningarna för korskopplingsreaktion och dessa organiska halogenider för att bilda kol-kolbindningar är kända som Suzuki-Miyaura-korskoppling .
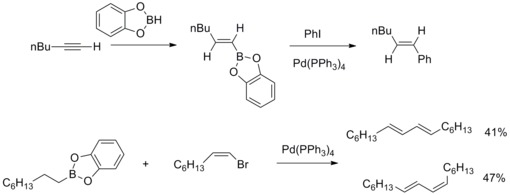
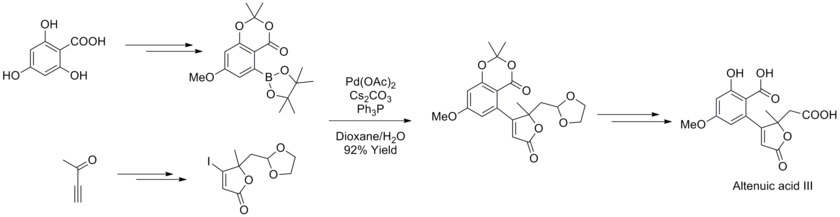
År 2013 bestämde Joachim Podlech och hans medarbetare strukturen av Alternaria mykotoxin altenuic acid III genom NMR-spektroskopisk analys och fullbordade dess totala syntes. I den syntetiska strategin användes Suzuki-Miyaura Cross-Coupling-reaktion med ett mycket funktionaliserat boronat och butenolider för att syntetisera en prekursor till den naturliga produkten med högt utbyte.
Modifierad Ullmann-biaryleter- och biarylaminsyntes
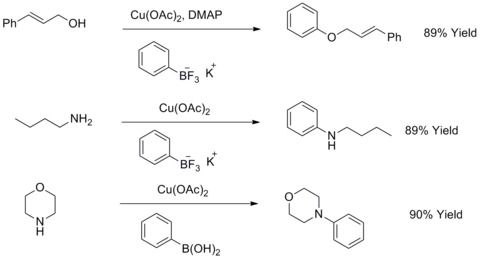
1904 fick Fritz Ullmann reda på att kopparpulver avsevärt kunde förbättra reaktionen mellan arylhalogenider och fenoler för att ge biaryletrar. Denna reaktion är känd som Ullmann-kondensation . År 1906 utökade I. Goldberg denna reaktion till att syntetisera en arylamin genom att reagera arylhalider med en amid i närvaro av kaliumkarbonat och CuI. Denna reaktion är känd som Goldberg-modifierad Ullmann-kondensation. 2003 har RA Batey och TD Quach modifierat denna typ av reaktioner genom att använda kaliumorganotrifluorboratsalter för att reagera med alifatiska alkoholer, alifatiska aminer eller aniliner för att syntetisera aryletrar eller arylaminer.
Se även
- Organoboron kemi
- Reaktioner av organoborater och boraner
- Corey–Itsuno-reduktion
- Midland Alpine boranreduktion
- Petasis reaktion
- Suzuki reaktion