PMS2
| PMS2 | |||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
 | |||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||
| Identifierare | |||||||||||||||||||||||||||||||||||||||||||||||
| , HNPCC4, PMS2CL, PMSL2, MLH4, PMS1 homolog 2, systemkomponent för felmatchning, MMRCS4, PMS-2 | |||||||||||||||||||||||||||||||||||||||||||||||
| Externa ID:n | |||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||
| Wikidata | |||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||



Mismatch reparation endonukleas PMS2 är ett enzym som hos människor kodas av PMS2 -genen .
Fungera
Denna gen är en av PMS2-genfamiljens medlemmar som finns i kluster på kromosom 7. Humana PMS2-relaterade gener är lokaliserade i banden 7p12, 7p13, 7q11 och 7q22. Exon 1 till 5 av dessa homologer delar hög grad av identitet med human PMS2. Produkten av denna gen är involverad i reparation av DNA-felmatchning . Proteinet bildar en heterodimer med MLH1 och detta komplex interagerar med MSH2 bundet till felmatchade baser. Defekter i denna gen är associerade med ärftlig nonpolypos kolorektal cancer, med Turcot syndrom , och är en orsak till supratentoriella primitiva neuroektodermala tumörer . Alternativt splitsade transkriptvarianter har observerats.
Mismatch reparation och endonukleasaktivitet
PMS2 är involverad i felparningsreparation och är känd för att ha latent endonukleasaktivitet som beror på integriteten hos det metabindande motivet i MutL-homologer. Som ett endonukleas introducerar PMS2 hack i en diskontinuerlig DNA-sträng.
Interaktioner
PMS2 har visats interagera med MLH1 genom att bilda heterodimeren MutLa. Det finns konkurrens mellan MLH3, PMS1 och PMS2 om den interagerande domänen på MLH1, som finns i resterna 492-742.
De interagerande domänerna i PMS2 har heptadupprepningar som är karakteristiska för leucinblixtlåsproteiner. MLH1 interagerar med PMS2 vid resterna 506-756.
MutS-heterodimererna, MutSa och MutSβ, associerar med MutLa vid felparningsbindning. MutLa antas förena felmatchningsigenkänningssteget med andra processer, inklusive: avlägsnande av felparningar från den nya DNA-strängen, återsyntes av det nedbrutna DNA:t och reparation av hacket i DNA:t. MutLa har visat sig ha svag ATPas-aktivitet och har även endonukleasaktivitet som introducerar hack i den diskontinuerliga DNA-strängen. Detta underlättar 5' till 3' nedbrytning av den felaktiga DNA-strängen av EXO1. Det aktiva stället för MutLa är beläget på PMS2-subenheten. PMS1 och PMS2 tävlar om interaktion med MLH1. Proteiner i interaktomen av PMS2 har identifierats genom tandemaffinitetsrening.
Human PMS2 uttrycks vid mycket låga nivåer och tros inte vara starkt cellcykelreglerad.
Interaktioner som involverar p53 och p73
PMS2 har också visat sig interagera med p53 och p73 . I frånvaro av p53 är PMS2-bristande och PMS2-kunniga celler fortfarande kapabla att stoppa cellcykeln vid G2/M-kontrollpunkten när de behandlas med cisplatin . Celler som har brist på p53 och PMS2, uppvisar ökad känslighet för anticancermedel. PMS2 är en skyddande mediator för cellöverlevnad i p53-bristceller och modulerar skyddsvägar för DNA-skador oberoende av p53. PMS2 och MLH1 kan skydda celler från celldöd genom att motverka p73-medierad apoptos på ett felaktigt reparationsberoende sätt.
PMS2 kan interagera med p73 för att förbättra cisplatin-inducerad apoptos genom att stabilisera p73. Cisplatin stimulerar interaktionen mellan PMS2 och p73, som är beroende av c-Abl. MutLa-komplexet kan fungera som en adapter för att föra p73 till platsen för skadat DNA och även fungera som en aktivator för p73, på grund av närvaron av PMS2. Det kan också vara möjligt för överuttryckt PMS2 att stimulera apoptos i frånvaro av MLH1 och i närvaro av p73 och cisplatin på grund av de stabiliserande effekterna av PMS2 på p73. Vid DNA-skada inducerar p53 cellcykelstopp genom p21 /WAF-vägen och initierar reparation genom uttryck av MLH1 och PMS2. MSH1/PMS2-komplexet fungerar som en sensor för omfattningen av skadan på DNA:t och initierar apoptos genom att stabilisera p73 om skadan inte kan repareras. Förlust av PMS2 leder inte alltid till instabilitet hos MLH1 eftersom det också kan bilda komplex med MLH3 och PMS1.
Klinisk signifikans
Mutationer
PMS2 är en gen som kodar för DNA-reparationsproteiner involverade i felmatchningsreparation . PMS2-genen finns på kromosom 7p22 och den består av 15 exoner. Exon 11 av PMS2-genen har en kodande upprepning av åtta adenosiner.
Omfattande genomisk profilering av 100 000 humana cancerprover visade att mutationer i promotorregionen av PMS2 är signifikant associerade med hög tumörmutationsbörda (TMB), särskilt vid melanom . TMB har visat sig vara en tillförlitlig prediktor för om en patient kan svara på cancerimmunterapi , där hög TMB är associerad med mer gynnsamma behandlingsresultat.
Heterozygota könslinjemutationer i DNA-felmatchningsreparationsgener som PMS2 leder till autosomalt dominant Lynch-syndrom. Endast 2% av familjer som har Lynch syndrom har mutationer i PMS2-genen. Patienternas ålder när de första gången fick PMS2-associerat Lynch-syndrom varierar mycket, med ett rapporterat intervall på 23 till 77 år.
I sällsynta fall kan en homozygot defekt orsaka detta syndrom. I sådana fall ärver ett barn genmutationen från båda föräldrarna och tillståndet kallas Turcot syndrom eller Constitutional MMR Deficiency (CMMR-D). Fram till 2011 har 36 patienter med hjärntumörer på grund av bialleliska PMS2 könslinjemutationer rapporterats. Nedärvning av Turcots syndrom kan vara dominant eller recessiv. Recessiv nedärvning av Turcots syndrom orsakas av sammansatta heterozygota mutationer i PMS2. 31 av 57 familjer som rapporterats med CMMR-D har mutationer i könslinje PMS2. 19 av 60 PMS2-homozygota eller sammansatta heterozygota mutationsbärare hade gastrointestinal cancer eller adenom som den första manifestationen av CMMR-D. Närvaro av pseudogener kan orsaka förvirring vid identifiering av mutationer i PMS2, vilket leder till falska positiva slutsatser om närvaron av muterad PMS2.
Brist och överuttryck
Överuttryck av PMS2 resulterar i hypermutabilitet och DNA-skadetolerans. Brist på PMS2 bidrar också till genetisk instabilitet genom att tillåta mutationer att föröka sig på grund av minskad MMR-funktion. Det har visat sig att PMS2-/- möss utvecklade lymfom och sarkom. Det visades också att hanmöss som är PMS2-/- är sterila, vilket indikerar att PMS2 kan ha en roll i spermatogenesen.
Roll i normal kolon
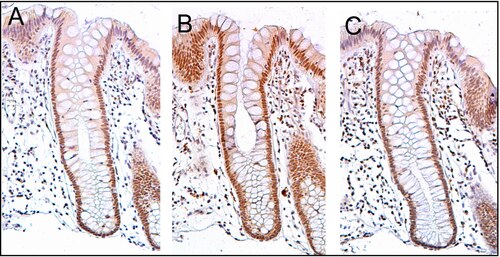
PMS2 uttrycks vanligtvis på en hög nivå i cellkärnor hos enterocyter (absorptiva celler) i tjocktarmskrypterna som kantar den inre ytan av tjocktarmen ( se bild, panel A). DNA-reparation, som involverar högt uttryck av proteiner PMS2, ERCC1 och ERCC4 (XPF), verkar vara mycket aktiva i tjocktarmskrypter i normalt, icke- neoplastiskt tjocktarmsepitel. När det gäller PMS2 är uttrycksnivån i normalt tjocktarmsepitel hög i 77 % till 100 % av krypterna.
Celler produceras vid kryptans bas och migrerar uppåt längs kryptaxeln innan de släpps in i kolonlumen dagar senare. Det finns 5 till 6 stamceller vid botten av krypterna. Om stamcellerna vid basen av kryptan uttrycker PMS2, kommer i allmänhet alla flera tusen celler i krypten också att uttrycka PMS2. Detta indikeras av den bruna färgen som ses av immunfärgning av PMS2 i de flesta enterocyterna i kryptan i panel A på bilden i detta avsnitt. Liknande uttryck av ERCC4 (XPF) och ERCC1 förekommer i tusentals enterocyter i varje kolonkrypta i det normala tjocktarmsepitelet.
Vävnadssektionen i bilden som visas här motfärgades också med hematoxylin för att färga DNA i kärnor med en blågrå färg. Cellkärnor i lamina propria (celler som finns under och omger epitelkrypterna) visar till stor del hematoxylin blågrå färg och har lite uttryck av PMS2, ERCC1 eller ERCC4 (XPF).
Koloncancer
Cirka 88% av celler av epitelialt ursprung i tjocktarmscancer och cirka 50% av tjocktarmskrypterna i epitelet inom 10 cm intill cancer (i fältdefekter från vilka cancerformerna troligen uppstod) har minskat eller frånvarande uttryck av PMS2.
Brister i PMS2 i kolonepitel verkar mest bero på epigenetisk repression. I tumörer som klassificeras som bristfällig och saknar reparation är PMS2-uttrycket i en majoritet bristfälligt på grund av bristen på sin parningspartner MLH1 . Parning av PMS2 med MLH1 stabiliserar. Förlusten av MLH1 i sporadiska cancerformer berodde på epigenetisk tystnad orsakad av promotormetylering i 65 av 66 fall. I 16 cancerformer var Pms2 bristfällig trots att MLH1-proteinuttryck var närvarande. Av dessa 16 fall fastställdes ingen orsak för 10, men 6 visade sig ha en heterozygot könscellsmutation i Pms2, följt av trolig förlust av heterozygositet i tumören. Således berodde endast 6 av 119 tumörer som saknade uttryck för Pms2 (5%) på mutation av PMS2.
Samordning med ERCC1 och ERCC4 (XPF)

När PMS2 reduceras i tjocktarmskryptor i en fältdefekt är det oftast associerat med minskat uttryck av DNA-reparationsenzymer ERCC1 och ERCC4 (XPF) också (se bilder i detta avsnitt). En brist på ERCC1 och/eller ERCC4 (XPF) skulle orsaka ansamling av DNA-skador. Sådan överflödig DNA-skada leder ofta till apoptos. En extra defekt i PMS2 kan dock hämma denna apoptos. Således skulle en ytterligare brist i PMS2 sannolikt väljas ut inför de ökade DNA-skadorna när ERCC1 och/eller ERCC4 (XPF) är bristfälliga. När ERCC1-bristiga äggstocksceller från kinesisk hamster upprepade gånger utsattes för DNA-skada, av fem kloner som härrörde från de överlevande cellerna, muterades tre i Pms2.
Progression till tjocktarmscancer
ERCC1, PMS2 dubbelmuterade äggstocksceller från kinesisk hamster, när de exponerades för ultraviolett ljus (ett DNA-skadande medel), visade en 7 375 gånger högre mutationsfrekvens än äggstocksceller från vildtyp kinesisk hamster och en 967 gånger högre mutationsfrekvens än de defekta cellerna i ERCC1, ensam. Således orsakar koloncellsbrist i både ERCC1 och PMS2 genominstabilitet . En liknande genetiskt instabil situation förväntas för celler som är dubbelt defekta för PMS2 och ERCC4 (XPF). Denna instabilitet skulle sannolikt öka progressionen till tjocktarmscancer genom att orsaka en mutatorfenotyp och förklara närvaron av cellerna som har dubbelt brist på PMS2 och ERCC1 [eller PMS2 och ERCC4 (XPF)] i fältdefekter associerade med tjocktarmscancer. Som indikerat av Harper och Elledge ligger defekter i förmågan att korrekt svara på och reparera DNA-skador bakom många former av cancer.
externa länkar
- Vanliga frågor om HNPCC Arkiverad 2007-08-15 på Wayback Machine från National Institute of Health
- GeneReviews/NCBI/NIH/UW-post om Lynch syndrom
|
PDB galleri
| |
|---|---|
|


