CAPP-Seq
| Akronym | CAPP-Seq |
|---|---|
| Används | Kvantifiering av lågnivå ctDNA från cancerpatienter. |
| Anmärkningsvärda experiment | CAPP-Seq applicerades på icke-småcellig lungcancer (NSCLC) för att identifiera återkommande somatiska förändringar från ctDNA. |
| Relaterade saker | Cellfritt tumör-DNA |
CA ncer P ersonaliserad profilering genom djup sekvensering (CAPP-Seq) är en nästa generations sekvenseringsbaserad metod som används för att kvantifiera cirkulerande DNA i cancer ( ctDNA ) . Metoden introducerades 2014 av Ash Alizadeh och Maximilian Diehns laboratorier på Stanford, som ett verktyg för att mäta Cellfritt tumör-DNA som frigörs från döda tumörceller till blodet och därmed kan spegla hela tumörgenomet. Denna metod kan generaliseras för alla cancertyper som är känd för att ha återkommande mutationer. CAPP-Seq kan detektera en molekyl mutant DNA i 10 000 molekyler friskt DNA. Den ursprungliga metoden förfinades ytterligare 2016 för ultrakänslig detektering genom integrering av flera felundertryckningsstrategier, så kallad integrerad Digital Error Suppression (iDES). Användningen av ctDNA i denna teknik bör inte förväxlas med cirkulerande tumörceller (CTCs); dessa är två olika enheter.
Ursprungligen beskrevs som en metod för att upptäcka och övervaka lungcancer, har CAPP-Seq framgångsrikt anpassats för ett brett spektrum av cancer av flera oberoende grupper. Dessa inkluderar diffust storcelligt B-cellslymfom (DLBCL), follikulärt lymfom (FL), post-transplantation lymfoproliferativ störning (PTLD), metastaserad kolorektal cancer i äggstockar, matstrupscancer , pankreascancer , blåscancer , leiomyosarkom , olika sarkom för vuxna och barn , bland andra.
Metod
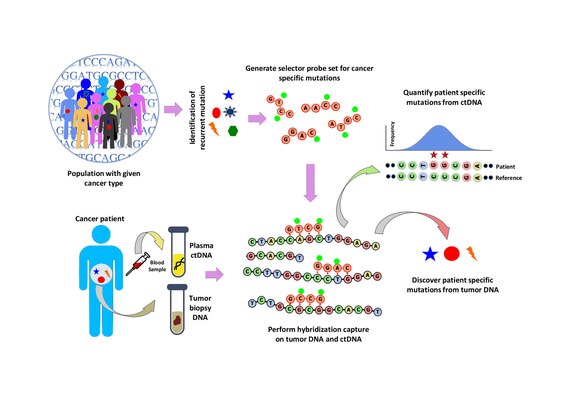
Populationsanalys utförs för att identifiera återkommande mutationer i en given cancertyp. Detta görs genom att analysera offentliga datamängder som COSMIC cancerdatabasen och TCGA . En "selektor" är utformad som består av biotinylerade DNA-oligonukleotidsonder riktade mot de återkommande muterade regionerna som valts för den specifika cancertypen. Väljaren väljs med hjälp av en flerfasig bioinformatisk metod. Med hjälp av väljaren utförs en sondbaserad hybridiseringsinfångning på tumör och normalt DNA för att upptäcka mutationer som är specifika för patienten. Hybridiseringsinfångningen appliceras sedan även på ctDNA för att kvantifiera de mutationer som tidigare upptäckts.
ctDNA-extraktion och biblioteksberedning
Perifert blod samlas in från patienter och ctDNA isoleras från ≥1 ml plasma . Inmatat DNA kan vara så lågt som 4 ng.
Det fanns fyra huvudmål med att anpassa detta protokoll för ctDNA-arbete:
- 1) för att optimera adapterligeringseffektiviteten
- 2) för att minska antalet PCR-cykler som behövs efter ligering
- 3) för att bevara den naturligt förekommande storleksfördelningen av ctDNA (median 170 baspar)
- 4) för att minimera variationen i djupet av sekvenstäckning över alla fångade regioner
Dessa uppnåddes genom att tillåta adapterligering att utföras vid 16 ℃ under 16 timmar för att öka adapterligeringseffektiviteten och återhämtningen. Den viktigaste anpassningen är under enzym- och saneringssteg; de utförs med pärla för att minimera röröverföringsstegen vilket ökar återhämtningen.
Selector design
I CAPP-Seq är utformningen av väljaren ett avgörande steg som identifierar återkommande mutationer i en viss cancertyp med hjälp av offentligt tillgängliga nästa generations sekvenseringsdata. För inkludering i CAPP-seq-selektorn beskrivs de återkommande mutationerna som är berikade i en population av ett index- Recurrence Index (RI). RI är antalet mutationer per kilobas av ett givet genomiskt lokus hos en patient som bär speciella mutationer. RI representerar en återfallsfrekvens på patientnivå som uppskattas för somatiska mutationer och alla mutationer. Kända och drivande återkommande mutationer i en population kan rangordnas baserat på RI och därför används RI för att designa en väljare. En designstrategi i sex faser används för att utforma väljaren.
- Fas-1: Identifiera ofta muterade kända drivrutinsmutationer med hjälp av allmänt tillgängliga data.
- Fas-2: Maximal täckning av SNV bland patienterna identifierades genom att rangordna deras exoniska RI.
- Fas-3 och 4: Exoner med högre RI valdes ut.
- Fas-5: Tillägg av tidigare förutsagda förarmutationer.
- Fas-6: Tillägg av återkommande genfusioner omarrangemang som är specifika för speciell cancer.
Human cancer är heterogen och återkommande cancermutationer förekommer endast i en minoritet av patienterna. Därför är en noggrann och icke-redundant design av väljaren den avgörande delen i CAPP-Seq och även storleken på väljaren är relaterad till dess nedströmskostnader.
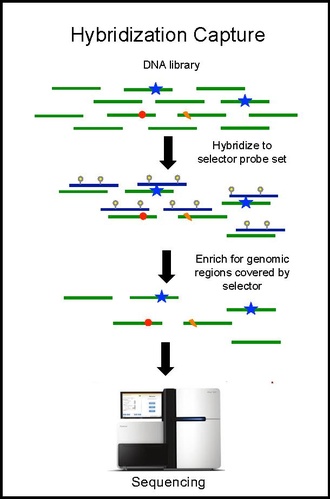
Hybridiseringsfångst och sekvensering
Hybridiseringsfångst med selektorprobsetet utförs på tumör-DNA från en biopsi och sekvenseras till ett djup av ~10 000× täckning. De biotinylerade selektorsonderna binder selektivt till de regioner i DNA-biblioteket som valdes att vara där de återkommande mutationerna inträffar i den givna cancertypen. På så sätt får du ett mindre bibliotek som är berikat för endast de regioner du vill ha, som sedan kan sekvenseras. Detta möjliggör bestämning av patientspecifika mutationer. Hybridiseringsinfångning med samma väljare utförs sedan på ctDNA från blodet för att kvantifiera de tidigare identifierade mutationerna hos patienten. CAPP-Seq kan appliceras på ctDNA från flera blodprover vid olika tidpunkter för att följa tumörutvecklingen.
Beräkningspipeline för CAPP-seq
En serie steg är involverade i analys av CAPP-Seq-data från mutationsdetektering till validering och öppen källkod kan göra det mesta av analysen. Efter det första steget av variantanrop tas könslinje- och förlust av heterozygositet (LOH) mutationer bort i CAPP-seq för att minska bakgrundsförändringarna. Flera statistiska signifikanstest kan utföras mot bakgrund av alla typer av variantanrop. Till exempel kan statistisk signifikans av tumörhärledda SNV: er uppskattas genom slumpmässig provtagning av bakgrundsalleler med Monte Carlo-metoden . För indel-anropen beräknas statistisk signifikans med användning av en separat metod som använde en strängspecifik analys med Z-test som visas i tidigare arbete. Slutligen minskar ett beräkningsvalideringssteg de falska positiva anropen. Ett robust beräkningsramverk specifikt för CAPP-seq-dataanalys är dock ett stort behov inom detta område.
Känslighet
Känsligheten hos denna teknik beror på den effektiva utformningen av väljaren och är mycket partisk med storleken på kohorten och typen av cancer som studeras. Bristen på bakgrund för att hitta de statistiskt signifikanta återkommande varianterna har begränsat dess prestanda på grund av stokastiskt brus och biologisk variabilitet. Receiver operating characteristic -analys (ROC) på flera cancerpatienter och cancerbotade patienter (prov insamlat vid olika tumörstadier, cirkulerande DNA-tidpunkt, behandling etc.) visade att CAPP-seq har högre sensitivitet och specificitet jämfört med tidigare metoder inom icke– småcellig lungcancer.
Begränsningar
Detektionsgränsen för CAPP-Seq påverkas av tre huvudområden: inmatad mängd ctDNA-molekyler, provkorskontaminering, potentiell allelbias i infångningsreagenset och PCR- eller sekvenseringsfel . ctDNA kan detekteras vid en nedre gräns på 0,025 % fraktionerad förekomst i blodet. Provkorskontaminering visade sig vara ett mycket litet bidrag och rapporter har visat minimal allelisk bias mot infångning av referensalleler i PBL ( perifera blodlymfocyter) . PCR- och sekvenseringsfel är också minimala. Tekniken blir tveksam när ctDNA är närvarande vid låga nivåer på 0,01 %. Dessutom, när det finns mindre utsläpp av ctDNA på grund av stabiliteten hos tumörtillväxt genom terapi, äventyras detektionen.
Huruvida ctDNA frisätts i samma eller olika hastighet från primära tumörer och metastaserande sjukdomar är fortfarande okänt. Detta faktum bör beaktas när man utför CAPP-Seq eftersom det kan orsaka problem med att bestämma tumörbördan och klonal utveckling om olika tumörer eller kloner dör ut och frisätter sitt DNA i olika hastigheter. Det är också okänt hur tumörhistologi påverkar ctDNA-frisättning.
En annan stor begränsning med att endast använda ctDNA-nivåer för att upptäcka tumörbörda är att ctDNA bara kan förutsäga kvarvarande tumör, det kan inte säga något om tumörens placering. Detta innebär att CAPP-Seq bäst kan användas som komplement till andra sekvenseringsmetoder för att avbilda sjukdomsbördan vid olika tidpunkter. Således är teknisk känslighet, reproducerbarhet, specificitet och krav på expertis för analys av stora mängder data några av de berörda frågorna med tekniken.
Fördelar
CAPP-Seq har många fördelar jämfört med andra metoder såsom digital polymeraskedjereaktion (dPCR) och amplikonsekvensering . CAPP-Seq kan kartlägga många loci i samma experiment jämfört med dPCR och amplikonsekvensering som använder flera olika experiment och därför använder mycket mer prov. En annan fördel är att CAPP-Seq inte bara kan upptäcka punktmutationer utan det kan också detektera indels , strukturella variationer och kopieringsnummervariationer och hjälper också till att övervaka minimal kvarvarande sjukdom.
En annan fördel med CAPP-Seq är att eftersom den bara riktar sig mot specifika områden av intresse i genomet är den mer kostnadseffektiv än hel exomsekvensering och helgenomsekvensering som är 171X respektive 44X dyrare. Dessutom finns det inget behov av diskret effektivisering för enskilda patienter.
Användning av cirkulerande tumör-DNA i motsats till solida tumörbiopsier möjliggör analys av hela repertoaren av tumörceller spridda genom tumören och avlägsna metastaser. Därför finns det en bättre chans att hitta alla mutationer associerade med denna cancer. Att ha en fullständig överblick över cancern och vad som driver den kommer att möjliggöra bättre behandlingsplaner och hantering av sjukdomen.
Ansökningar
Övervakning av tumörbördan
Vid behandling av cancer är det användbart att ha exakta mätningar av den totala sjukdomsbördan i kroppen. Det hjälper till att bestämma prognostisk signifikans och behandlingssvar. Detta görs normalt med datortomografi ( CT-skanningar ), positronemissionstomografi ( PET-skanningar ) eller magnetisk resonanstomografi ( MRT ). Dessa medicinska bildbehandlingsförfaranden är dyra och är inte utan sina egna problem. Dessa avbildningstekniker kan inte exakt lösa små tumörer (≤1 cm i diameter). Avbildning kan också påverkas av strålningsinducerad inflammation och fibrotiska förändringar, vilket gör det svårt att avgöra om det finns kvarvarande tumör eller bara effekter av behandlingen.
Det har visat sig att nivåer av ctDNA i plasma signifikant korrelerar med tumörvolymen jämfört med medicinsk bildbehandling (CT, PET och MRI). Detektering av ctDNA kan förutsäga kvarvarande tumör eller förestående återfall, i vissa fall till och med bättre än medicinsk bildbehandling och nuvarande metoder.
Prognostisk indikator
Detektion av ctDNA har hittills visat sig vara en prediktor för återfall i flera studier. I en studie i sent stadium av NSCLC (icke-småcellig lungcancer) fann de två fall där ctDNA korrekt bestämde resultatet för en patient när medicinsk bildbehandling var felaktig. I ett fall förutspådde bildtagningen återfall baserat på en misstänkt kvarvarande tumör som visade sig endast vara strålningsinducerad inflammation, men ctDNA upptäcktes inte och patienten återkom inte. I ett annat fall visade avbildningen ingen tumör men ctDNA upptäcktes och patienten fick återfall kort därefter. I en annan studie på DLBCL (diffust stort B-cellslymfom) visade sig ctDNA också vara förutsägande för återfall.
Biopsifri tumörgenotypning
Biopsier är invasiva och förknippade med risker för patienten. Därför är flera biopsier för att övervaka sjukdomsprogression sällsynta och diagnostiska biopsier förlitas på för genetisk information. Detta kan vara problematiskt på grund av tumörheterogenitet och tumörutveckling. För det första provar biopsier bara en del av tumören, och eftersom tumörer är heterogena kommer detta inte att täcka hela tumörens genetiska landskap. För det andra, efter behandling utvecklas tumörer och det kan finnas nya mutationer som inte är representerade i det diagnostiska provet.
Biopsifri tumörgenotypning, med hjälp av CAPP-Seq och ctDNA, tar upp många av dessa problem. Ett enkelt blodprov är icke-invasivt och mycket säkrare och lättare att utsätta cancerpatienter för flera gånger under behandlingsförloppet. Att använda ctDNA ger ett bättre prov av tumör-DNA jämfört med ett enda område av en tumör som samlats in i en biopsi, vilket möjliggör en bättre uppskattning av tumörheterogenitet. Genom att ta flera prover av ctDNA vid olika tidpunkter efter behandlingsförloppet kan tumörutvecklingen avslöjas. Detta kan hjälpa till att upptäcka uppkomsten av mutationer som ger resistens mot en riktad terapi och tillåta behandlingsförloppet att anpassas därefter. CAPP-Seq möjliggör specifikt screening av flera genomiska platser, vilket kommer att bli viktigt när listan över cancermutationer som är viktiga för behandling fortsätter att växa. I en studie för sent stadium av NSCLC utförde de en version av CAPP-Seq där tumörbiopsien inte sekvenserades först, och de kunde korrekt klassificera 100 % av patientens plasmaprover med en 0 % falsk positiv frekvens. Detta visar att även utan tidigare kunskap om tumörmutationer, kan de upptäckas exakt med enbart ctDNA.