Chip-exo
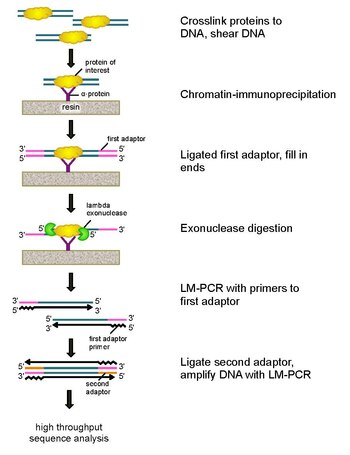
ChIP-exo är en kromatinimmunoutfällningsbaserad metod för att kartlägga de platser där ett protein av intresse ( transkriptionsfaktor ) binder till genomet. Det är en modifiering av ChIP-seq -protokollet, som förbättrar upplösningen av bindningsställen från hundratals baspar till nästan ett baspar. Den använder användningen av exonukleaser för att bryta ned strängar av det proteinbundna DNA:t i 5'-3'-riktningen till ett litet antal nukleotider från proteinbindningsstället. Nukleotiderna i de exonukleasbehandlade ändarna bestäms med användning av någon kombination av DNA-sekvensering , mikroarrayer och PCR . Dessa sekvenser mappas sedan till genomet för att identifiera platserna på genomet där proteinet binder.
Teori
Chromatin immunoprecipitation ( ChIP ) tekniker har använts sedan 1984 för att detektera protein-DNA-interaktioner. Det har funnits många varianter av ChIP för att förbättra kvaliteten på resultaten. En sådan förbättring, ChIP-on-chip (ChIP-chip), kombinerar ChIP med mikroarrayteknik. Denna teknik har begränsad känslighet och specificitet, särskilt in vivo där mikroarrayer är begränsade av tusentals proteiner som finns i det nukleära utrymmet, vilket resulterar i en hög frekvens av falska positiva. Därefter kom ChIP-sequencing (ChIP-seq), som kombinerar ChIP med high-throughput-sekvensering. Den heterogena naturen hos klippta DNA-fragment kartlägger emellertid bindningsställen till inom ±300 baspar, vilket begränsar specificiteten. För det andra utgör kontaminerande DNA ett allvarligt problem eftersom så få genetiska loci är tvärbundna till proteinet av intresse, vilket gör varje icke-specifikt genomiskt DNA till en betydande källa till bakgrundsljud.
För att lösa dessa problem reviderade Rhee och Pugh den klassiska nukleasskyddsanalysen för att utveckla ChIP-exo. Denna nya ChIP-teknik förlitar sig på ett lambda- exonukleas som bara bryter ned, och allt, obundet dubbelsträngat DNA i 5′-3′-riktningen. Kortfattat tvärbinds ett protein av intresse (utformning av ett med en epitoptagg kan vara användbart för immunoutfällning) in vivo till dess naturliga bindningsplatser över ett genom med användning av formaldehyd. Celler samlas sedan upp, bryts upp och kromatinet klipps och solubiliseras genom sonikering . En antikropp används sedan för att immunoutfälla proteinet av intresse, tillsammans med det tvärbundna DNA:t. DNA PCR-adaptrar ligeras sedan till ändarna, som tjänar som en startpunkt för syntes av andra strängens DNA efter exonukleas-digestionen. Lambda-exonukleas smälter sedan dubbla DNA-strängar från 5′-änden tills digestion blockeras vid gränsen för protein-DNA-kovalent interaktion. Mest kontaminerande DNA bryts ned genom tillsats av ett andra enkelsträngsspecifikt exonukleas. Efter att tvärbindningen har reverserats, förlängs primrarna till PCR-adaptrarna för att bilda dubbelsträngat DNA, och en andra adapter ligeras till 5'-ändar för att avgränsa den exakta platsen för exonukleasuppslutningsupphörande. Biblioteket amplifieras sedan med PCR, och produkterna identifieras genom sekvensering med hög genomströmning . Denna metod möjliggör upplösning av upp till ett enda baspar för vilket proteinbindningsställe som helst inom vilket genom, vilket är en mycket högre upplösning än antingen ChIP-chip eller ChIP-seq.
Fördelar
ChIP-exo har visat sig ge upp till en enda basparsupplösning vid identifiering av proteinbindningsplatser. Detta är i motsats till ChIP-seq som kan lokalisera ett proteins bindningsställe endast med ±300 baspar.
Kontaminering av icke-proteinbundna DNA-fragment kan resultera i en hög andel falska positiva och negativa i ChIP-experiment. Tillsatsen av exonukleaser till processen förbättrar inte bara upplösningen av bindningsställeanrop, utan tar bort kontaminerande DNA från lösningen före sekvensering.
Proteiner som är ineffektivt bundna till ett nukleotidfragment är mer benägna att detekteras av ChIP-exo. Detta har till exempel möjliggjort igenkänning av fler CTCF-transkriptionsfaktorbindningsställen än vad som tidigare upptäckts.
På grund av den högre upplösningen och minskade bakgrunden behövs mindre sekvenstäckning när du använder ChIP-exo.
Begränsningar
Om ett protein-DNA-komplex har flera platser för tvärbindning inom en enda bindningshändelse, kan det se ut som om det finns flera distinkta bindningshändelser. Detta beror sannolikt på att dessa proteiner denatureras och tvärbinds vid ett av de tillgängliga bindningsställena inom samma händelse. Exonukleaset skulle sedan stanna vid ett av de bundna ställena, beroende på vilket ställe proteinet är tvärbundet till.
Som med alla ChIP-baserade metoder måste en lämplig antikropp för proteinet av intresse vara tillgänglig för att kunna använda denna teknik.
Ansökningar
Rhee och Pugh introducerar ChIP-exo genom att utföra analyser på en liten samling transkriptionsfaktorer: Reb1, Gal4, Phd1, Rap1 i jäst och CTCF i människa. Reb1-platser hittades ofta i kluster och dessa kluster hade ~10 gånger högre beläggning än förväntat. Sekundära ställen i kluster hittades ~40 bp från ett primärt bindningsställe. Bindningsmotiv av Gal4 visade en stark preferens för tre av de fyra nukleotiderna, vilket tyder på en negativ interaktion mellan Gal4 och den uteslutna nukleotiden. Phd1 känner igen tre olika motiv vilket förklarar tidigare rapporter om tvetydigheten i Phd1:s bindningsmotiv. Rap1 visade sig känna igen fyra motiv. Ribosomala proteingener bundna av detta protein hade en tendens att använda ett speciellt motiv med en starkare konsensussekvens. Andra gener använde ofta kluster av svagare konsensusmotiv, möjligen för att uppnå en liknande beläggning. Bindande motiv av CTCF använde fyra "moduler". Hälften av de bundna CTCF-ställena använde modul 1 och 2, medan resten använde någon kombination av de fyra. Man tror att CTCF använder sina zinkfingrar för att känna igen olika kombinationer av dessa moduler.
Rhee och Pugh analyserade pre-initiation complex (PIC) struktur och organisation i Saccharomyces genom. Med hjälp av ChIP-exo kunde de, bland andra upptäckter, exakt identifiera TATA-liknande egenskaper i promotorer som rapporterats vara TATA-lösa.